Хромосомные болезни, или синдромы - это группа врожденных патологических состояний, проявляющихся множественными пороками развития, различающихся по своей клинической картине, часто сопровождающихся тяжелыми нарушениями психического и соматического развития. Основной дефект - различные степени интеллектуальной недостаточности, что может осложняться нарушениями зрения, слуха, опорно-двигательного аппарата, более выраженными, чем интеллектуальный дефект, расстройствами речи, эмоциональной сферы и поведения.
Диагностические признаки хромосомных синдромов можно разделить на три группы:
неспецифические, т.е. такие, как выраженная умственная отсталость, сочетающаяся с дисплазиями, врожденными пороками развития и черепно-лицевыми аномалиями;
признаки, характерные для отдельных синдромов;
патогномоничные для конкретного синдрома, например, специфический плач при синдроме «кошачьего крика».
Хромосомные заболевания не подчиняются менделеевским закономерностям передачи заболевания потомству и в большинстве случаев обнаруживаются спорадически, являясь следствием мутации в половой клетке одного из родителей.
Хромосомные болезни могут быть унаследованы, если мутация имеется во всех клетках родительского организма.
К механизмам, лежащим в основе геномных мутаций, относятся:
нерасхождение - хромосомы, которые должны были разделяться во время клеточного деления, остаются соединенными и относятся к одному полюсу;
«анафазное отставание» - утрата отдельной хромосомы (моносомия) может иметь место во время анафазы, когда одна хромосома может отстать от остальных;
полиплоидизация - в каждой клетке геном представлен более чем дважды.
Факторы, повышающие риск рождения детей с хромосомными болезнями
Причины возникновения хромосомных болезней до настоящего времени недостаточно изучены. Имеются экспериментальные данные о влиянии на мутационный процесс таких факторов, как: действие ионизирующих излучении, химических веществ, вирусов. Другими причинами нерасхождения хромосом могут быть: сезонность, возраст отца и матери, порядок рождения детей, прием лекарств во время беременности, гормональные нарушения, алкоголизм и др. Не исключается до определенной степени и генетическое детерминирование нерасхождения хромосом. Повторим, однако, что причины образования геномных и хромосомных мутаций на ранних стадиях развития зародыша до сих пор окончательно не раскрыты.
К биологическим факторам повышения риска рождения детей с хромосомными аномалиями может быть отнесен возраст матери. Риск рождения больного ребенка особенно резко возрастает после 35 лет. Это характерно для любых хромосомных болезней, но наиболее четко наблюдается для болезни Дауна.
В медико-генетическом планировании беременности особое значение уделяется двум факторам - наличию анеуплоидии по аутосомам у ребенка и возрасту матери старше 35 лет.
К кариотипическим факторам риска у супружеских пар относятся: анеуплоидия (чаще в мозаичной форме), робертсоновские транслокации (слияние двух телоцентрических хромосом в области деления) кольцевые хромосомы, инверсии. Степень повышения риска зависит от типа хромосомных нарушений.



Синдром Дауна (трисомия по 21 паре хромосом)
Причина: Нерасхождение 21 пары аутосом, транслокация 21 аутосомы на аутосому группы D или G. У 94% кариотип - 47 хромосом. Частота проявления синдрома увеличивается с возрастом матери.
Клиника: Признаки, позволяющие диагностировать заболевание, в типичных случаях выявляются на самых ранних этапах жизни ребенка. Малый рост ребенка, маленькая круглая голова со скошенным затылком, своеобразное лицо - бедная мимика, косой разрез глаз со складкой у внутреннего угла, нос с широкой плоской переносицей, маленькие деформированные ушные раковины. Рот обычно полуоткрыт, язык толстый, неповоротливый, нижняя челюсть иногда выступает вперед. На щеках часто отмечается сухая экзема. Обнаруживается укорочение конечностей, особенно в дистальных отделах. Кисть плоская, пальцы рук широкие, короткие. В физическом развитии отстают, однако не резко, но нервно-психическое развитие замедленно (плохо развита речь). С возрастом выявляется ряд новых черт заболевания. Голос грубеет, отмечается близорукость, косоглазие, конъюнктивиты, неправильный рост зубов, кариес.Слабо развита иммунная система, инфекционные заболевания протекают крайне тяжело и в 15 раз чаще, чем у других детей. Встречается острый лейкоз.
Патогенез: Патологии внутренних органов, сердечно-сосудистые дефекты.
Диагностика: Клиническое обследование, подтверждаемое цитогенетическим анализом кариотипа.
Лечение: Комплексная терапия, включающая правильную организацию режима, рационально построенная медико-педагогическая работа, лечебная физкультура, массаж, медикаментозное лечение.


Синдром Тернера-Шершевского (ХО)
Причина: Нерасхождение половых хромосом, отсутствие одной Х-хромосомы, кариотип - 45 хромосом.
Клиника : Низкий рост,непропорциональное строение тела, полная короткая шея с крыловидными кожными складками, широкая грудная клетка, Х-образное искривление коленей. Уши деморфированы, низко расположены. Отмечается неправильный рост зубов. Половой инфантилизм. Снижение умственного развития.
Патогенез: В пубертатный период недоразвитие половых органов и вторичных половых признаков, поражение сосудистой системы, аномалии мочевой системы, уменьшение остроты зрения, слуха.
Диагностика : У новорожденных ее установить трудно. С возрастом диагностика основывается на клинической картине и определении патологии кариотипа и полового хроматина.
Лечение: Симптоматическое, направленное на увеличение роста. Для увеличения роста используются анаболические гормоны. С 13-15 лет начинают лечение эстрогенными препаратами. Полного выздоровления не наблюдается, однако лечебные мероприятия могут улучшить состояние


Синдром Клайнфельтера (XXY ; XYY ; XYYYY ; XXXY )
Причина: Нерасхождение половых хромосом, вследствие чего увеличивается число X или Y хромосом в клетке, кариотип - 47 (XXY), 48 и более хромосом.
Клиника: Высокий рост, отсутствие залысин на лбу, плохой рост бороды, гинекомастия, остеохондроз, бесплодие, слаборазвиты мышцы, аномалия зубов и костной системы. Больные могут демонстрировать сниженный интеллект. С увеличением X-хромосом увеличивается умственная отсталость до полной идиотии, с увеличением Y-хромосом - агрессивность. Больные с более глубокой степенью интеллектуального дефекта могут обнаруживать ряд психопатологических признаков: они мнительны, склонны к алкоголизму, способны совершать различные правонарушения.
Патогенез: В пубертатном периоде обнаруживается недоразвитие первичных половых признаков.
Диагностика: Основана на клинических данных, а также на определении патологического кариотипа цитогенетическим методом, что подтверждается исследованием полового хроматина в клетках.
Лечение: Терапия с помощью мужских половых гормонов для увеличения потенции. Психотерапия.

Синдром Волъфа-Хиршхорна
Причина: У 80 % страдающих им новорожденных цитологическую основу данного синдрома составляет деления короткого плеча 4-й хромосомы. Размеры делеции колеблются от небольших терминальных до занимающих около половины дистальной части короткого плеча. Отмечается, что большинство делеции возникает заново, около 13 % происходит, в результате транслокаций у родителей. Реже в геноме больных, помимо траснлокации, имеются и кольцевые хромосомы. Наряду с делениями хромосом, патология у новорожденных может быть обусловлена инверсиями, дупликациями, изохромосомами.
Клиника: У новорожденных небольшой вес при нормальной продолжительности беременности. Также отмечаются микроцефалия, клювовидный нос, эпикант, антимонголоидный разрез глаз (опущение наружных углов глазных щелей), аномальные ушные раковины, расщелина верхней губы и неба, маленький рот, деформация стоп и др. Дети с синдромом Вольфа-Хиршхорна маложизнеспособны, как правило умирают в возрасте до одного года.
Патогенез: Болезнь характеризуется многочисленными врожденными пороками развития, задержкой умственного и психомоторного развития.
Диагностика: По клинической картине.
Лечение: Не существует.
Синдром трисомии (XXX )
Причина: Нерасхождение половых хромосом в результате нарушения работы митотического веретена деления во время мейоза, кариотип - 47 хромосом.
Клиника: Пузырное нерасхождение плаценты; новорожденный имеет небольшой, широкий задний родничок, недоразвитые затылочные и теменные кости черепа. Отставание в развитии на 6-7 месяцев. Низко расположены деформированные ушные раковины. Синдактилия пальцев кисти, расщелина губы и неба, гидроцефалия. Многие женщины нормально развиты, интеллект ниже среднего. Частота развития шизофреноподобных психозов увеличивается второе.
Патогенез: Пороки развития внутренних органов.
Диагностика: По клинической картине и цитогенетическому определению патологии кариотипа и полового хроматина.
Лечение: Симптоматическое.

Синдром Эдвардса (трисомия по 18 паре хромосом)
Причина: Нерасхождение аутосом на стадии гамет (иногда зигот). Лишняя хромосома в 18 паре. Кариотип 47, Е18+. Выражена зависимость частоты рождения больных детей от возраста родителей.
Клиника: Пренатальное недоразвитие, слабая активность плода, нарушения строения лица (короткие глазные щели, маленькая верхняя челюсть) и костно-мышечной системы практически постоянны. Ушные раковины деформированы и в подавляющем большинстве случаев расположены низко. Грудина короткая, ядра окостенения расположены неправильно и в меньшем количестве. Спинномозговые грыжи и расщелины губ.
Патогенез: Наиболее постоянны пороки сердца и крупных сосудов. Нарушения развития головного мозга, в основном гипоплазия мозжечка и мозолистого тела. Из пороков глаз чаще всего обнаруживается микроанафтольмия. Врожденное отсутствие щитовидной железы и надпочечников.
Диагностика: Клинический осмотр, дерматоглифика,
цитогенетическое обследование.
Лечение: Отсутствует, 90% детей умирают на первом году жизни. Выжившие дети умирают от инфекционных заболеваний, чаще от пневмонии.


Синдром Патау (трисомия но 13 таре аутосом)
Причина: Нерасхождение аутосом 13 пары в гаметогенезе у одного из родителей. Кариотип - 47, D13+ .
Клиника: Аномалии черепа и лица, окружность черепа обычно уменьшена, в ряде случаев имеется выраженная тригоноцефалия. Умеренная микроцефалия сочетается со сравнительно низким и скошенным лбом, узкими глазными щелями, запавшим предносьем с широким основанием носа, низко расположенными и деформированными ушными раковинами. Расстояние между глазными щелями часто уменьшено. На коже головы имеются дефекты скальпа овальной или круглой формы. Часто – заячья губа и волчья пасть. Аномалии костно-мышечной системы, полидактилия.
Патогенез: Смертность в течение первого года жизни (90%). Основной причиной смерти детей являются тяжелые, несовместимые с жизнью пороки развития: дефекты сердечно-сосудистой и мочеполовой систем, аномалии толстой кишки, пупочная грыжа, нарушения строения глазных яблок, постоянны микроанофтальмия, дисплазия сетчатки, катаракты. Врожденные пороки сердца встречаются у 80% детей.
Диагностика: Основана на клиническом, цитогенетическим исследованиях.

Синдром "кошачьего крика"
Причина: Делеция короткого плеча хромосомы 5-й пары. Кариотип 46, 5р-.
Клиника: Патологическое строение голосовых связок - сужение, мягкость хрящей, отечность и необычная складчатость слизистой, мяуканье кошки. Недоразвитие речи. Микроцефалия. Лунообразное лицо, монголоидный разрез глаз, косоглазие, катаракта, атрофия зрительного нерва, плоская спинка носа, высокое нёбо, деформированные ушные раковины. Косолапость. Задержка умственного и физического развития. Продолжительность жизни значительно снижена, только около 14% больных переживают возраст 10 лет.
Патогенез: Порок сердца.
Диагностика: Клиническое обследование с выявлением наиболее постоянного признака синдрома - "кошачий крик", дерматоглифика и цитогенетическое выявление патологии кариотипа.
Лечение: Отсутствует.

Синдром Орбели
Причина : Деления длинного плеча аутосомы 13.
Клиника: Лоб переходит в нос, не образуя носовой вырезки. Большое расстояние между глазами. Широкая спинка носа, высокое нёбо, низко расположенные диспластичные ушные раковины, пороки развития глаз (косоглазие, катаракта). Пороки опорно-двигательного аппарата -неспецифические аномалии (косолапость, вывих тазобедренных суставов). Задержка роста и психомоторного развития; характерна глубокая олигофрения. Больные с развернутой клинической картиной синдрома погибают на первом году жизни.
Патогенез: Аномальное развитие практически всех органов и систем; микроцефалия; врожденные пороки сердца и аномалии прямой кишки.
Диагностика:
Лечение: Отсутствует.
Синдром Мориса
Причина: Мутация гена, нарушающая образование нормального белка - рецептора, делает ткани-мишени невосприимчивому гормону, направляющему их развитие по мужскому типу. Не использовав такую возможность на определенном этапе онтогенеза, организм осуществляет развитие по женскому типу.
Клиника: Появляется особь с кариотипом XY, но внешне более сходна с женщиной. Такие субъекты не способны иметь потомство, так как их половые железы (семенники) недоразвиты, а их выводные протоки часто формируются по женскому типу (недоразвитая матка, влагалище). Вторичные половые признаки также характерны для женского пола.
Патогенез: Недоразвитые половые органы.
Диагностика: Цитогенетическое, клиническое обследование.
Лечение: Гормональная терапия.

Бывает 45 хромосом. Если 47 хромосом- это синдром Дауна, то интересно, бывает ли 45 хромосом и получил лучший ответ
Ответ от Ёельдерея Сельдеревна))[гуру]
Да. Синдром Шерешевского-Тернера, например. Моносомия по Х хромосоме
Ответ от 2 ответа
[гуру]
Привет! Вот подборка тем с ответами на Ваш вопрос: Бывает 45 хромосом. Если 47 хромосом- это синдром Дауна, то интересно, бывает ли 45 хромосом
Ответ от Виталик максимович
[гуру]
Ответ от Вадим Рустамович Абдрахимов
[активный]
Ответ от хавани
[эксперт]
Ответ от Виталик максимович
[гуру]
конечно в этом мире всё бывает.. .
Ответ от Вадим Рустамович Абдрахимов
[активный]
бывает, "Отсутствие в хромосомном наборе диплоидного организма одной хромосомы называется моносомией (2n-1); отсутствие двух гомологичных хромосом - нуллисомией (2n-2); наличие дополнительной хромосомы называется трисомией (2n+1) ."
Ответ от хавани
[эксперт]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Yamaha yamaha
[новичек]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Pepsimonk / RORSCHAH
[активный]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Дима Лемкин
[новичек]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Anna Kushnirenko
[новичек]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Иерт Ерок
[новичек]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Марсель шарипов
[новичек]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Eisenhorn
[активный]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Їестимир Четвертак
[новичек]
по идеи у человека должны быть 46, если хромосомы выше или ниже то выделяется у человека какая-то болезнь.
Ответ от Леонид Гулько
[новичек]
Одни спецы по избытку хромосом.
Хромосомные болезни - это большая группа врожденных наследственных болезней, вызываемых аномалиями в количестве или структуре хромосом, то есть мутациями. Заболевания, которые вызываются геномными и хромосомными мутациями, называются хромосомными болезнями .
Патологические изменения возникают как при потере генетического материала, так и при добавлении новых хромосом.
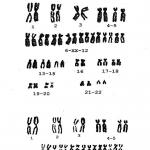
Заболевания, вызываемые мутациями аутосом (Рис. 1):
Рис. 1. Болезни, вызываемые мутациями аутосом ()
Делеция в 5-й хромосоме вызывает синдром кошачьего крика, у новорожденных больных отмечается нарушение строения гортани, мяукающий тембр голоса, слабоумие, отсталость психомоторики, такие больные редко доживают до зрелого возраста;
Делеция в 3-й хромосоме, как правило, приводит к прерыванию беременности, при рождении дети не способны сидеть и есть твердую пищу;
Делеция в 21-й хромосоме вызывает хроническое белокровие, нехватку красных кровяных телец;
Трисомия по 21-й хромосоме (болезнь Дауна) (Рис. 2) - в кариотипе у больных не две, а три 21-х хромосомы - это самая распространенная аномалия, частота рождения составляет 1:500, зависит от возраста матери и резко возрастает после 35 лет. До 40 % детей с такой болезнью рождаются у возрастных матерей.

Рис. 2. Синдром Дауна ()
У таких больных наблюдается монголоидный тип лица, укороченные конечности, психическая отсталость.
Трисомия по 13-й хромосоме (Синдром Патау) - довольно редкий синдром, частота возникновения 1:14 500, у больных аномалии сердца и почек, серьезные нарушения внешности, продолжительность жизни не более года;
Трисомия по 18-й хромосоме (синдром Эдвардса) - у больных множественные пороки органов, умственная отсталость, смертность в раннем возрасте.
Встречаются трисомии по 8-й, 9-й, 14-й и 22-й хромосоме, они все летальны на раннем этапе.
Существует единичное описание даже тетросомии (умственная отсталость разной степени) и пентосомии (тяжелые поражения организма и умственных способностей) аутосом.

Рис. 3. Болезни, связанные с нарушениями половых хромосом у женщин ()
Трисомия ХХХ (синдром трипло-Х) - частота возникновения 1:700, не резкие отклонения в физическом развитии, нарушения функции яичников, преждевременный климакс. Больные с такой мутацией даже не догадываются о своем кариотипе;
Тетросомия ХХХХ приводит к умственной недостаточности разной степени;
Пентосомия ХХХХХ сопровождается тяжелыми повреждениями органов и сознания;
Моносомия Х0 (синдром Тернера) - единственная совместимая с жизнью, частота возникновения 1:4000, недоразвитые яичники и матка, физическая и умственная отсталость.
- синдром Клайнфельтера встречается в двух формах - полисомия по Х-хромосоме и полисомия по Y-хромосоме, больные с кариотипом ХХ Y - это мужчины женоподобного типа, у них развита грудь, женский голос, длинные ноги, недоразвиты семенники. Они бесплодны, но психически совершенно нормальны;

Рис. 4. Болезни, связанные с нарушениями половых хромосом у мужчин ()
Больные с кариотипом Х Y Y - нормальные высокие мужчины, умственно и психически здоровы, но склонны к агрессии, асоциальны.
Приведенные мутации не вызывают гибель организма, прочие, как правило, приводят к гибели еще на этапе внутриутробного развития. Нарушения, вызванные этими патологиями, обычно не совместимы с половым процессом, такие больные не доживают до репродуктивного возраста или являются стерильными. Поэтому хромосомные болезни, в отличие от генных, гораздо хуже передаются по наследству, но есть и исключение - больные с синдромом Дауна способны к размножению.
В результате интенсивного изучения хромосом человека и хромосомных болезней на протяжении 35-40 лет сложилось учение о хромосомной патологии, которая имеет большое значение в современной медицине.
Список литературы
- Беляев Д.К. Общая биология. Базовый уровень. - 11 издание, стереотипное. - М.: Просвещение, 2012.
- Пасечник В.В., Каменский А.А., Криксунов Е.А. Общая биология, 10-11 класс. - М.: Дрофа, 2005.
- Агафонова И.Б., Захарова Е.Т., Сивоглазов В.И. Биология 10-11 класс. Общая биология. Базовый уровень. - 6-е изд., доп. - Дрофа, 2010.
- Bibliofond.ru ().
- Rh-conflict.narod.ru ().
- Vse-pro-geny.ru ().
Домашнее задание
- Дать определение хромосомным болезням.
- Каковы основные заболевания, вызываемые мутациями аутосом?
- От чего зависит вероятность рождения ребенка с синдромом Дауна?
Патологии половых хромосом могут быть обусловлены нарушением их количества (анеуплоидиями) или же структурными дефектами.
Наиболее распространенные анеуплоидии половых хромосом: 45,X (Синдром Тёрнера); 47,XXY (Синдром Клайнфельтера); 47,XYY; и 47,XXX. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом - 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует доле аномальных клеток.
Структурные патологии X- и Y-хромосом прежде всего включают изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Одним из примеров геномного расстройства является дупликация гена MECP2 у мужчин, выражающаяся в наличии мышечной гипотонии, тяжелой умственной отсталости, задержки речевого развития, нарушения глотания, частых респираторных инфекций, а также судорожных приступов (тонико-клонические судороги, не поддающихся лечению).
Аномалии числа хромосом (анеуплоидии)
Наиболее частыми анеуплоидиями половых хромосом являются 45,X (Синдром Шерешевского-Тернера); 47,XXY (Синдром Клайнфельтера); 47,XYY и 47,XXX с частотой возникновения приблизительно 1/2500, от 1/500 до 1/1000, от 1/900 до 1500 и 1/1000 соответственно. Мозаицизм по половым хромосомам с присутствием в организме клеток с нормальным генотипом нередок. Два наиболее распространенных вида мозаицизма половых хромосом - 45,X/46,XX и 45,X/46,XY. Тяжесть фенотипических проявлений у пациентов с мозаицизмом соответствует проценту аномальных клеток.
Моносомия по X-хромосоме (45,X, или Синдром Шерешевского-Тёрнера)
Большинство пациентов с синдромом Шерешевского-Тёрнера имеют моносомию по Х-хромосоме, кариотип 45,X. Другие формы синдрома включают мозаицизм по хромосоме Х, например, 45,X/46,XX или 45,X/46,XY с частичной делецией Y-хромосомы. У некоторых пациентов имеется структурная аномалия второй X-хромосомы (например, изохромосомия длинного плеча X-хромосомы или делеция короткого плеча). Делеции, включающие в себя дистальную часть короткого плеча Y-хромосомы, также ассоциированы с фенотипом синдрома Тёрнера, поскольку в данном случае у пациентов отсутствуют так называемые анти-тёрнеровские гены (SHOX, RPSY4 и ZFY). Делеции короткого плеча X-хромосомы также связывают с фенотипом синдрома Тёрнера. В большинстве представляют собой единичные случаи.
Синдром Шерешевского-Тёрнера характеризуется низкорослостью и некоторыми из следующих проявлений: дисморфия лица, включающая низко посаженные уши, кожные складки на шее, щитообразная грудная клетка (широкая, с большим расстоянием между сосками), лимфедема, вальгусная деформация локтевого сустава, короткая четвертая пястная кость, гипоплазия ногтевых пластин, пигментные пятна и врожденные пороки сердца. Среди пороков сердца типичными и наиболее часто встречающимся являются дефекты сосудов и коарктация аорты. Вдобавок у пациентов, страдающих синдромом Тёрнера, развиваются полосковидные гонады, наблюдается нарушение овуляции и задержка полового развития. Также встречаются дефекты развития почек (подковообразная почка). Лимфедема нижних отделов конечностей может быть единственным клиническим признаком, наблюдаемым у новорожденных. Лица с синдромом Тёрнера, несущие генетический материал Y-хромосомы, имеют повышенный риск развития гонадобластомы.
47,XXY Синдром Клайнфельтера
Синдром Клайнфельтера является самой распространенной патологией числа половых хромосом, вызывающей первичный гипогонадизм. Кариотип 47,XXY является результатом нерасхождения половых хромосом и может быть как материнским, так и отцовским по происхождению. Большинство случаев болезни обнаруживается постнатально и диагностируется при определении причин бесплодия, выявлении гинекомастии, крипторхизма или же неврологических нарушений.
Рис. Нерасхождение половых хромосом
Новорожденные мальчики с кариотипом 47,XXY фенотипически нормальны, с физиологически нормальными мужскими наружными половыми органами и без какой-либо видимой дисморфии. Основные клинические проявления синдрома Клайнфельтера, включающие высокий рост, маленькие яички и бесплодие (азооспермия), становятся выраженными в постпубертатном периоде. У пациентов с синдромом Клайнфельтера повышен риск психических расстройств, расстройств аутистического характера и социальных проблем. У пациентов с диагностированным синдромом Клайнфельтера следует оценивать неврологический статус и направлять к эндокринологу.
47,XYY
Лица с кариотипом 47,XYY имеют высокий рост, у них может отмечаться умеренная задержка в двигательном и речевом развитии. Для многих из них требуется повышенное внимание к обучению, но, как правило, все они учатся в основных общеобразовательных школах. Половое развитие проходит нормально и большинство мальчиков фертильны. Из-за невыраженности фенотипа и отсутствия связанных с этим проблем со здоровьем, многие лица с кариотипом 47,XYY на протяжении всей их жизни остаются недиагностированными.
Ранее сообщалось, что у мужчин с 47,XYY повышена агрессия, что выражается в их агрессивном поведении. Однако последующие крупномасштабные совместные исследования европейских и американских генетиков показали, что статистика повышенной криминальной деятельности мужчин с XYY коррелировала с их низким социально-экономическим статусом по причине низкого значения IQ (около 10 баллов), что приводило к определенным трудностям с законом и, чаще, незначительным правонарушениям. У лиц с 47,XYY отмечаются более высокие показатели синдрома дефицита внимания и гиперактивности, а также расстройств аутистического характера. Таким пациентам рекомендуется оценка их нервно-психического развития, учитывая широкую распространённость трудностей в обучении и поведенческих проблем.
47,XXX
47,XXX (она же трисомия по X-хромосоме) является самой распространенной патологией половых хромосом у женщин. Трисомия по Х-хромосоме диагностируется внутриутробно в ходе генетического скрининга. У женщин с кариотипом 47,XXX нет повышенного риска развития плода с хромосомными аномалиями.
Обследование 155 женщин с кариотипом 47,XXX показало, что 62 процента из них были физически нормальными. Таким образом, для большинства лиц с кариотипом 47,XXX диагноз никогда не устанавливается. У женщин с 47,XXX отмечается высокий рост; (средняя длина окружности головы варьирует в пределах 25 - 35 процентиль, однако к подростковому возрасту для многих может достигать 80 процентиль). Половозрелость и фертильность чаще всего в норме, но может отмечаться преждевременное угасание функции яичников.
В следующем обследовании одиннадцати младенцев с кариотипом 47,XXX было показано, что коэффициент интеллекта девочек с рождения был на 15-20 баллов ниже, чем у их братьев. Поэтому рекомендуется отслеживать задержки в развитии и выявлять наличие психологических проблем в дальнейшем.
Другие заболевания
Сообщалось о более чем ста случаях кариотипа 49,XXXXY, по меньшей мере двадцати случаях 49,XXXXX и нескольких - 49,XYYYY. Прослеживается прямая зависимость между числом дополнительных половых хромосом и тяжестью фенотипических проявлений у пациентов. В исследовании тетра- и пентасомии половых хромосом сделан вывод о том, что полисомия по X-хромосоме связана с более тяжкими последствиями, чем полисомия по Y-хромосоме. Было показано, что уровень интеллекта IQ снижается на 10 пунктов с каждой лишней X-хромосомой от их нормального числа.
49,XXXXY Характерными клиническими чертами кариотипа XXXXY являются запавшая переносица с широким или приподнятым кончиком носа, широко расположенные глаза, веко-носовые складки, скелетные патологии (особенно лучелоктевой синостоз), врожденные сердечные заболевания, эндокринные расстройства и высокая степень гипогонадизма и гипогенитализма. Также обычным являются выраженная умственная отсталость и умеренная низкорослость. Хотя лиц с таким кариотипом часто относят к случаям синдрома Клайнфельтера, все характерные черты XXXXY довольно отчетливо указывают именно на данный фенотип.
49,XXXXX У женщин с кариотипом 49,XXXXX (пентасомия по X-хромосоме) всегда присутствует умственная отсталость. Другие проявления, такие как черпено-лицевые, сердечно-сосудистые и скелетные патологии довольно непостоянны. У пациентов, страдающих пентасомией по X-хромосоме, могут проявляться схожие черты с теми, что наблюдаются при синдроме Дауна. Лучелоктевой синостоз также часто выражен у пациентов с большим числом X-хромосом. Некоторые пациенты имеют мозаицизм 48,XXXX и 49,XXXXX.
Мозаицизм 45,X/46,XX
Это наиболее распространенный мозаицизм половых хромосом, который диагностируется при амниоцентезе и пренатальном кариотипировании. У лиц с данным типом мозаицизма имеются более легкие клинические черты синдрома Тёрнера. Многие женщины прошли половое созревание и смогли воспроизвести потомство.
Из 156 пренатально диагностированных случаев мозаицизма 45,X/46,XX 14% случаев имели ненормальный исход. Было зарегистрировано два мертворождения и 20 случаев ненормального фенотипа (у 12 имелись некоторые черты синдрома Тёрнера, а остальные 8 носили характер аномалий, возможно, не связанных с ним). Более 85 % девочек имели нормальный фенотип при рождении, либо он был установлен по результатам медицинского прерывания беременности. Однако, главные черты синдрома Тёрнера (такие как низкий рост и отсутствие вторичных половых признаков) проявились только в детстве или юности, и не были замечены в младенчестве. У некоторых женщин с нормальным фенотипом, при нарушении функции яичников, выявляется мозаицизм 45,X/46,XX.
Мозаицизм 45,X/46,XY
Мозаицизм с наличием 45,X/46,XY имеет широкий фенотипический спектр. Например, в ретроспективной серии 151 постнатально диагностированных случаев мозаицизма 45,X/46,XY, 42 % пациентов - девочки по фенотипу, с наличием типичного или нетипичного синдрома Тёрнера. Еще у 42 % наблюдались неопределённые наружные половые органы и асимметричные гонады (смешанный гонадный дисгенез), наконец, у 15% был мужской фенотип с неполной маскулинизацией. Таким образом, все случаи, диагностированные постнатально, были фенотипически патологичными. Напротив, среди 80 пренатально диагностированных случаев мозаицизма 45,X/46,XY 74 92,6% были нормальными по фенотипу мальчиками. Это может объяснить тот факт, что дети или взрослые с наличием мозаицизма, но нормальным фенотипом вряд ли стали бы обращаться за медицинской помощью (ошибка обращаемости).
Структурные аномалии хромосом
Структурные патологии включают, прежде всего, изохромосомы, делеции, дупликации, кольцевые хромосомы и транслокации.
Изохромосома Xq
Изохромосома длинного плеча X-хромосомы, isoXq или i(Xq), при наличии которой короткое плечо (p) исключено (отсутствует/редуцировано) и заменено точной копией длинного плеча (q), - является наиболее распространенной аномалией половых хромосом.
Наличие структурной патологии не связывают с повышенным возрастным риском родителей. Изохромосомия 46,X,i(Xq) может быть выражением мозаицизма, когда в организме присутствуют две генетически разные клеточные популяции: нормальная - 46,XX и 45,X.
Изохромосомы Xq и Xy ассоциируют с синдромом Тёрнера, возможно, потому, что главный анти-тёрнеровский ген SHOX располагается на дистальной части коротких плеч X-и Y-хромосом (на псевдоаутосомных областях). Изохромосома Xq также выявляется у пациентов в одной из вариаций синдрома Клайнфельтера, 47,X,i(Xq),Y.
Делеция Xp22.11
Делеция Xp22.11 включает в себя ген PTCHD1 . Сообщалось о выявлении в нескольких семьях с расстройствами аутистического характера, а также в трёх семьях с умственной отсталостью. Ген PTCHD1 является геном-кандидатом в отношении Х-сцепленной умственной отсталости, проявляющейся с аутизмом или без аутизма. Функция и роль данного гена неизвестны.
Делеция Xp22.3
Делеция данной области часто ассоциируется с синдромом микрофтальмии и линейных дефектов кожи (MLS) и является Х-сцепленным доминантным нарушением, то есть, летальным для мужчин и поэтому прослеживающимся только у женщин. Ген в данной области кодирует митохондриальную цитохром-c-синтазу (HCCS ). Клиническое проявление MLS выражается наличием микрофтальмии и анофтальмии (одно- или двусторонней) и линейными дефектами кожи, в основном лица и шеи, которые со временем проходят. Структурные патологии головного мозга, задержка в развитии и приступы (припадки) тоже входят в состав клинической картины. Нарушения сердечной деятельности (как гипертоническая кардиомиопатия и аритмия), низкий рост, грыжа диафрагмы, ногтевая дистрофия, преаурикулярный свищ, потеря слуха, мочеполовые мальформации (пороки развития, неправильное формирование) также являются частыми клиническими явлениями.
Скрининговая оценка предусматривает офтальмологический и дерматологический осмотр, оценку общего развития, выполнение эхокардиограммы, магнитно-резонансной томографии мозга (МРТ) и электроэнцефалограммы (ЭЭГ).
Делеции Xp22 SHOX
Делеция Xp22 включает в себя ген SHOX, мутация которого является причиной идиопатического низкого роста. Ген SHOX находится в псевдоаутосомном регионе 1 X- и Y-хромосом. Этот ген считается ответственным за низкорослость при синдроме Тёрнера, а гаплонедостаточность данного гена вызывает дисхондростеоз Лери-Вейлля. Дисхондростеоз Лери-Вейлля характеризуется низким ростом, наиболее выражено проявляющимся у женщин, а также хроническим подвывихом кисти (деформацией костей запястья, деформация Маделунга). Гомозиготные делеции гена SHOX вызывают дисплазию Лангера, более тяжелую форму метафизарной дисплазии. Делеции гена SHOX легко обнаруживаются у пациентов с низким ростом, без каких-либо других специфических особенностей в строении их скелета. Более чем 60% перестроек SHOX - это делеции гена; при отсутствии делеций сравнительная геномная гибридизация с последующим секвенированием для выявления и установления точечных мутаций, является клиническим обследованием идиопатического низкого роста.
Делеции Xp11.22
Делеции региона Xp11.22 включают ген PHF8 (кодирует пальцевидный белок PHD8), мутации которого связывают с умственной отсталостью, наличием расщелины губы/неба, а также с расстройствами аутистического характера.
Мутации с делецией гена PHF8 ассоциированы с синдромом Х-сцепленной умственной отсталости, синдром Сидериус-Хамель (синдром Siderius-Hamel).
Дупликации Xp.22.31
Дупликации в локусе Xp.22.31 часто описываются в литературе. Было много дискуссий на тему того, является ли данная дупликация патогенетической или же доброкачественным явлением, учитывая трудности определения последствий вариации числа копий генов. Данная дупликация затрагивает ген стероидной сульфатазы. Как результат - генетический дефект, мутация в гене стероидной сульфатазы, что выражается в снижении её активности или отсутствие её синтеза. Делеция данного гена связана с Х-сцепленным ихтиозом у мужчин. Данная дупликация отмечается у пациентов с умственной отсталостью. Однако, она выявляется как у здоровых родственников этих пациентов, так и в основной популяции. Хотя дупликации данного гена могут и не иметь фенотипических проявлений, трипликации последовательно связывают с умственными расстройствами. FISH-диагностика позволяет в конечном счете дифференцировать дупликации от трипликации (распознать увеличение копийности гена).
Синдром дупликации ME2CP
Мутации в гене, кодирующем метил-связывающий-CpG терминальный белок 2 (ME2CP ), расположенный в Xq28, ответственный за синдром Ретта. Дупликации данного региона имеет небольшое или вовсе не имеет фенотипического значения для женщин, вероятно, из-за инактивации патологической X-хромосомы. Мужчины с данной мутацией сильно ослаблены. Наличие дупликации клинически выражается в наличии выраженной мышечной гипотонии, тяжелой умственной отсталости, задержке речевого развития, нарушения глотания (трудностей приема пищи), частых респираторных инфекций и судорожных приступов вплоть до тонико-клонических, иногда не поддающихся лечению. Многие пациенты с наличием данной дупликации были с диагностированным аутизмом либо расстройством подобного типа. По аналогии с тем, что наблюдается в синдроме Ретта, пациенты с дупликацией ME2CP испытывают регресс развития. Вдобавок у них развивается атаксия, прогрессирующие мышечные спастичности нижней части тела часто приводят к потере способности передвигаться. Отмечались проблемы желудочно-кишечного тракта и сильные запоры. Дупликация часто затрагивает ген антагонист рецептора интерлейкина 1 (IRAK1 ), что может играть роль в появлении иммунных патологий, отмечаемой у данной группы пациентов. Прогноз неблагоприятен, и большинство мужчин с данной дупликацией умирают до 30 лет по причине вторичных респираторных инфекций. Трипликация данного региона проявляется еще более тяжелым фенотипом у мужчин.
Скрининговые обследования этих пациентов предполагают проведение ЭЭГ, оценку функции глотания, оценку гуморального и клеточного иммунитета. Лечение может включать лечение мышечной гипотонии и спастичности, речевую терапию (логопедию), использование гастрономической трубки (гастростома) в случае проблем с питанием, а также лечение респираторных инфекций.
Перевод материалов сайта UpTodate подготовлен специалистами Центра иммунологии и репродукции.
Хромосомные болезни – недуги наследственного характера, которые обуславливаются изменением структуры или количества хромосом. К этой группе заболеваний также относятся те, которые вызываются геномными мутациями. Возникают патологии вследствие изменений, происходящих в половых клетках родителей.
Понятие о хромосомных болезнях
Это большая группа врожденных недугов, которая занимает одно из лидирующих мест в списке наследственных патологий человека. Цитологические исследования материалов ранних абортов показывают, что хромосомоные болезни человека могут проявляться еще у эмбрионов. То есть, заболевания развиваются еще в процессе оплодотворения или на ранних этапах дробления зиготы.
Виды хромосомных болезней
Специалисты привыкли разделять все недуги на три больших вида. Классификация хромосомных болезней зависит от нарушений:
- плоидности;
- количества хромосом;
- структуры хромосом.
Самые распространенные аномалии, вызванные нарушением плоидности – триплодия и тетраплодия. Такие изменения, как правило, фиксируются только в материале, получаемом в результате абортов. Известны лишь единичные случаи рождения детей с подобными нарушениями, и они всегда мешали нормальной жизнедеятельности. Триплодия – результат оплодотворения диплоидных яйцеклеток гаплоидными сперматозоидами или наоборот. Иногда аномалия является последствием оплодотворения одной яйцеклетки двумя сперматозоидами.
Нарушение числа хромосом

В большинстве случаев хромосомные болезни, к которым привело нарушение числа хромосом, проявляются целой моносомией или трисомией. При последней все три нуклеопротеидные структуры являются гомологами. При первой аномалии числа хромосом одна из двух имеющихся в наборе остается нормальной. Целая моносомия бывает только по хромосоме Х, поскольку эмбрионы с другими наборами погибают очень рано – еще на начальных этапах внутриутробного развития.
Нарушение структуры хромосом
Недуги, развивающиеся на фоне нарушения структуры хромосом, представлены большой группой синдромов с частичной моно- или трисомией. Возникают они, когда имеют место структурные изменения в родительских половых клетках. Такие нарушения влияют на процессы рекомбинации. Из-за этого в мейозе происходит утрата или переизбыток фрагментов нуклеопротеидных структур. Частичные хромосомные аномалии могут наблюдаться в любых хромосомах.
Причины хромосомных болезней
Ученые долгое время работали над этим вопросом. Как выяснилось, хромосомные мутации болезни вызывают. Они приводят к отклонениям в строении и функциях нуклеопротеидных структур. Знать нужно не только причины возникновения хромосомных болезней, но и факторы, располагающие к проявлению мутаций. Значение имеют:
- особенности участвующей в аномалии;
- генотип организма;
- тип аномалии;
- размеры недостающего или избыточного генетического материала (при структурных нарушениях);
- степень клеточной мозаичности организма (в расчет берутся только те клетки, у которых есть отклонения в строении или функциях).
Хромосомные болезни – список
Каждый год он пополняется новыми названиями – недуги исследуются постоянно. Рассматривая, какие есть хромосомные болезни, самые известные сегодня это:
- Синдром Дауна. Развивается по причине трисомии. То есть, потому, что в клетках присутствует три экземпляра 21-й хромосомы, вместо двух. Как правило, «лишняя» структура передается новорожденному от матери.
- Синдром Клайнфельтера. Эта хромосомная болезнь проявляется не сразу при рождении, а только после полового созревания. В результате данного отклонения мужчины получают от одной до трех Х-хромосом и теряют возможность иметь детей.
- Миопия. является генетическим отклонением, из-за которого изображение формируется не там, где положено – на сетчатке глаза, – а перед нею. Главная причина этой проблемы – увеличение глазного яблока в длину.
- Дальтонизм. Дальтоники не могут различать один или сразу несколько цветов. Причина – в «дефектной» хромосоме Х, получаемой от матери. У представителей сильного пола данное отклонение встречается чаще, потому как у мужчин Х-структура только одна, и «исправить дефект» – как то происходит в случае с женскими организмами – их клетки не могут.
- Гемофилия. Хромосомные болезни могут проявляться и нарушением свертываемости крови.
- Мигрень. Заболевание, проявляющееся сильными болями в голове, тоже передается по наследству.
- Муковисцидоз. Для этого недуга характерно нарушение работы желез внешней секреции. Люди с таким диагнозом страдают от повышенной потливости, обильного отделения слизи, скапливающейся в организме и мешающей корректной работе легких.
Методы диагностики хромосомных болезней

Генетические консультации, как правило, обращаются за помощью к таким методам:
- Генеалогический. Заключается в сборе и анализе данных о родословной пациента. Этот метод позволяет понять, в самом ли деле болезнь наследственная и если так, определить тип наследования.
- Антенатальной диагностики. Определяет наследственные нарушения плода, который находится в утробе матери на сроке 14-16 недель беременности. Если в околоплодной жидкости выявляются аномалии аутосом, может быть сделан .
- Цитогенетический. Используется для выявления синдромов и отклонений.
- Биохимический. Уточняет болезни и помогает выявить мутированные гены.
Лечение хромосомных болезней
Терапия не всегда помогает избавиться от недуга, но может замедлить его течение. Хромосомные аномалии плода лечатся такими методами:
- Диетотерапия. Предполагает добавление в рацион или исключение из него определенных веществ.
- Медикаментозная терапия. Применяется с целью повлиять на механизмы синтеза ферментов.
- Хирургическое лечение. Помогает справиться с , разными костными дефектами и деформациями.
- Заместительная терапия. Суть ее – в возмещении тех веществ, которые самостоятельно в организме не синтезируются.
Частота хромосомных болезней
Очень часто хромосомные аномалии человека встречаются в материалах, полученных в результате спонтанных абортов, сделанных в первом триместре. Общая частота нарушений в популяции на самом деле не так велика и составляет около 1%. Дети с генетическими отклонениями могут рождаться и у здоровых родителей. Новорожденные девочки и мальчики, как показывает медицинская практика, поражаются хромосомными болезнями с одинаковой частотой.